Neural-Network-Based Selective Configuration Interaction Approach to Molecular Electronic Structure
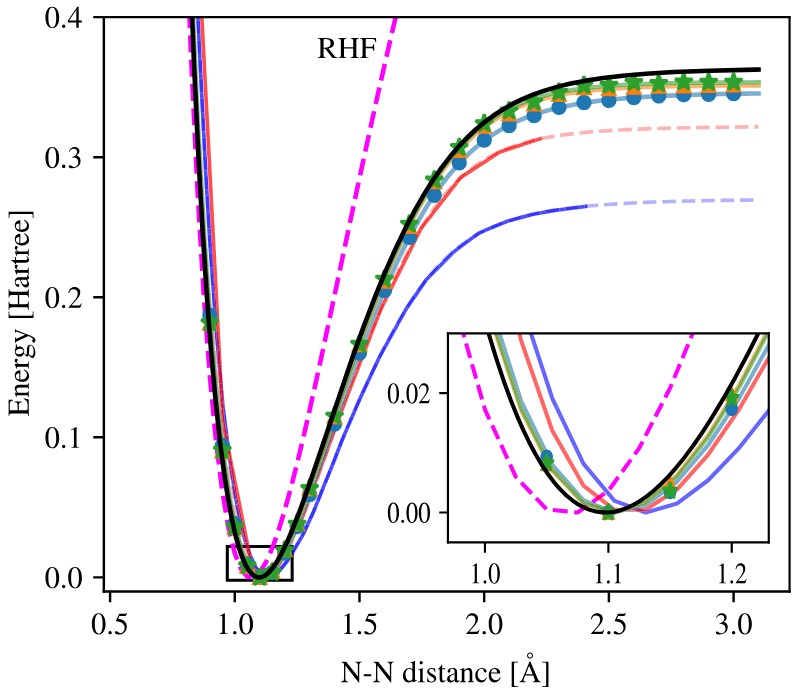
We are pleased to announce our new joint publication with Theoretical Chemsits at the University of Iceland in the Journal of Chemical Theory and Computation (JCTC). In this work, we combine Hartree–Fock with a neural-network-supported quantum-cluster solver to introduce the Neural-Network Configuration Interaction (NNCI) method for accurate and efficient electronic-structure calculations. NNCI selectively identifies relevant Slater determinants via a neural classifier and active learning, enabling near full-CI accuracy with drastically fewer determinants.
The method is benchmarked on several small molecules and achieves excellent agreement with established CI calculations—reproducing correlation energies for the N₂ molecule using only (4 \times 10^5) determinants compared to (10^{10}) in conventional CI. Its efficiency and scalability open a path toward embedding applications and the study of larger molecular and condensed-matter systems.
Authors: Yorick L.A. Schmerwitz, Louis Thirion, Gianluca Levi, Elvar Ö. Jónsson, Pavlo Bilous, Hannes Jónsson, and Philipp Hansmann
Published: J. Chem. Theory Comput. 21 (5), 2025
DOI: https://doi.org/10.1021/acs.jctc.4c01234