Our Research
We study how interacting electrons give rise to surprising states of matter and how modern machine learning can help solve long-standing quantum problems. Our main research branches are:
Correlated Adatoms on Surfaces
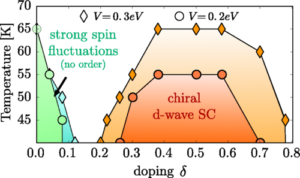
We investigate transition-metal adatoms placed on well-controlled semiconductor surfaces (such as SiC). These atomic-scale systems are ideal playgrounds for strong electronic correlations: local moments, Kondo physics, flat bands, and interaction-driven phases can all appear, sometimes within a single platform. By combining model Hamiltonians (Anderson/Hubbard), advanced numerical methods, and material-realistic simulations, we ask: Which atomic arrangements and substrates stabilize magnetism, Mott insulating behavior, or even superconductivity?
This line of research connects fundamental many-body theory with experimentally accessible real material platforms. Beyond explaining observed phenomena, we aim to design correlated surface architectures with targeted low-energy properties—opening routes to quantum functionality in engineered materials.
Neural-Network Configuration Interaction (NNCI) with Quantum Chemistry
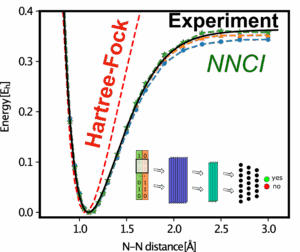
In collaboration with Dr. Pavlo Bilous (MPL), we develop Neural-Network Configuration Interaction (NNCI) to tackle the exponential complexity of molecular electronic structure. NNCI augments established wave-function approaches (e.g., CI and Hartree–Fock) with machine-learned representations that efficiently capture the most relevant electron configurations while preserving the physical rigor of the CI approach.
Working closely with also with collaborators in theoretical chemistry (In the group of Prof. Hannes Jónsson at the University of Iceland), we apply and validate NNCI on molecular benchmarks such as N₂ (see plot of the N2 dissociation curve) and are now extending it to larger hydrocarbons like propane. The current generation of our method surpasses state-of-the-art Full Configuration Interaction (FCI) benchmarks, achieving chemical accuracy correlation energies at a fraction of the computational cost. This marks a significant step toward scalable many-electron solutions that bridge quantum chemistry and condensed matter, demonstrating that neural-network–enhanced quantum solvers can now reach — and even exceed — the accuracy of the world’s best conventional electronic structure methods.
Our long term goal are accurate calculations for heterogeneous catalysis.
SOLAX — Neural-Network-Supported Configuration Interaction Code

SOLAX is our open-source code package that integrates neural networks into configuration-interaction (CI) calculations. It provides a flexible, modular framework for constructing and optimizing many-electron wave functions with machine-learned components while preserving the physical interpretability of the CI approach. Within SOLAX, neural networks act as adaptive basis optimizers and configuration selectors, enabling accurate descriptions of strongly correlated systems at a fraction of the traditional computational cost. The code has been benchmarked on molecular systems such as N₂ and serves as the computational backbone for our ongoing development of the Neural-Network Configuration Interaction (NNCI) method. SOLAX is designed for transparency and extensibility, facilitating reproducible research and cross-disciplinary applications between condensed-matter and quantum-chemistry communitie